BACKGROUND
Cutis laxa syndrome (CL) is a rare connective tissue disorder characterized by hyperextensible and inelastic loose hanging skin. Redundant skin leads to excessive skin folds in the whole body but mostly prominent around eyes, face, neck and hands giving a premature ageing appearance. (1)
The condition is caused by degenerative changes in the elastic fibers and may be inherited or acquired. The inherited forms may be autosomal dominant due to heterozygous variants in the elastin (ELN) gene or recessive and X-linked recessive (multiple genes have been reported including FBLN5, ALDH18A1, PYCR1, LTBP1, LTBP4, EFEMP, ATP6V1A, ATP6V0A2) although the recessive form has most commonly been reported. (2)
Elastic fibers provide elasticity and resilience to skin, lungs, eyes, and large blood vessels. CL can have variable systemic manifestations ranging from mild to severe, some of them can be present at birth such as inguinal hernia and joint hyperlaxity and others evolve during childhood. (3)
In terms of cardiovascular involvement, various abnormalities have been reported including aortic dilatation, pulmonary artery branch stenoses and right-sided heart failure generally caused by pulmonary disease. (4)
INTRODUCTION
We present a case of a 5-year-old with genetically confirmed CL. She was born at term via uncomplicated spontaneous vaginal delivery. She was the only child of unrelated parents.
She had antenatal suspicion of renal pelvis dilatation, however several postnatal scans showed stable and mild degree dilatation with otherwise normal morphology.
On examination at birth, she had multiple folds of excess skin on her whole body especially her face, neck, and trunk. (Figure 1.) She had normal hair, mucosa, conjunctiva, nails, and eyes. No evidence of hypermobility and she had normal tone.
She underwent genetic testing that showed a change in the elastin gene. She was found to have likely pathogenic heterozygous c.2098dup, p. (Arg700fs) in ELN gene. This result along with her phenotype was consistent with the diagnosis of autosomal dominant CL.
Her father also had similar skin changes. He was one of seven siblings and no other family members showed a similar phenotype. He was found to have aortic valve disease and had an aortic valve replacement in his 30s. He sadly died ten years shortly before our patient’s birth due to sepsis after redo aortic surgery. He never had genetic testing, his presentation is described by Roussin et al. (5)

FIRST ASSESSMENT
When A first attended clinic she was growing and thriving and had no development delay. She was not on any medication and had no allergies. At 18-month-old she was diagnosed with bilateral inguinal hernia and underwent uncomplicated surgical repair.
She was assessed by the respiratory team and had a normal chest X-ray with no respiratory concern. She had regular surveillance with dermatology and ophthalmology.
From the cardiac point of view there were no clinical concerns with no floppy episodes or any other symptoms. She was in Ross functional class 1 with no exercise limitation.
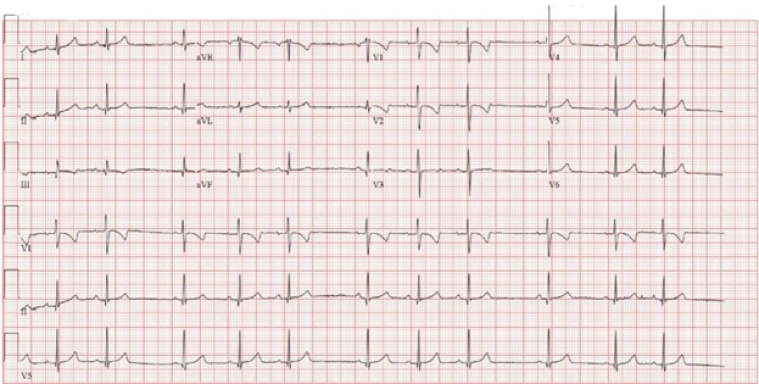
Her ECG showed sinus rhythm with no atrio-ventricular and intraventricular conduction delay and normal repolarization pattern of repolarization for her age. (figure 2)
IMAGING
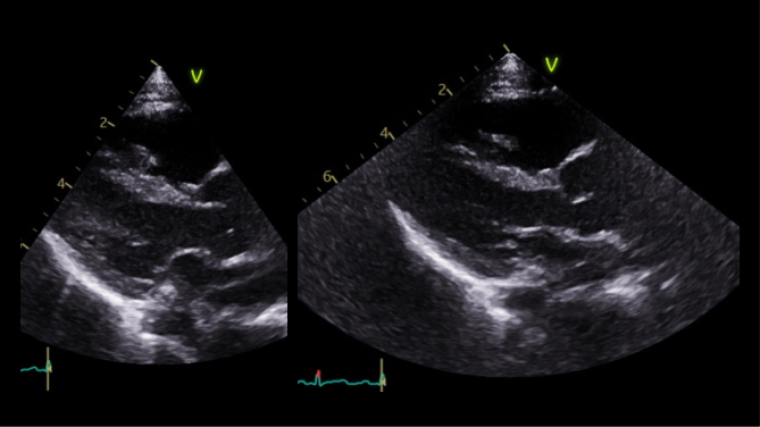
The first echocardiogram was performed when she was 8-month-old showing normal intracardiac anatomy and dimension, mild central aortic regurgitation and a moderate dilatation of the aorta with aortic annulus 13 mm (Z score +0.30), sinus of Valsalva 23.3 mm (Z score +3.71), ST junction 19.2 mm (Z score +3.48) and ascending aorta 19.1 mm (Z score +2.41), Lopez dataset.
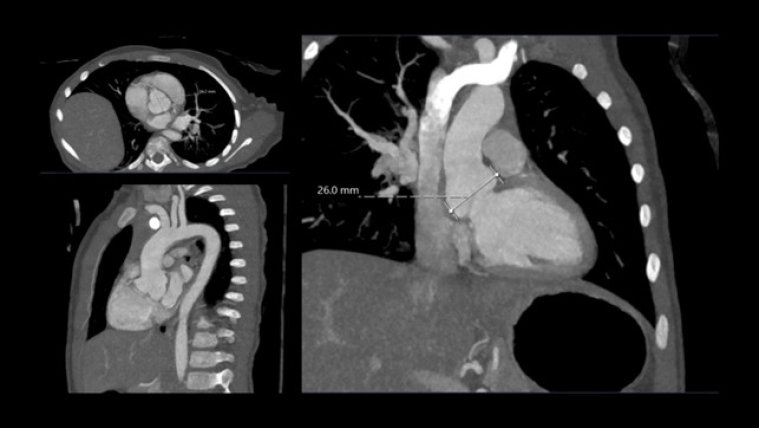
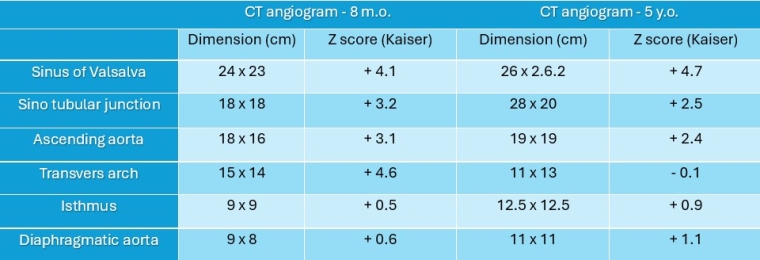
A month later she underwent cross sectional imaging. Her CT angiogram head to pelvis showed the aortic root was dilated and the thoracic aorta was elongated at the ascending and arch level, and tortuous with rightwards convexity in the descending portion. Z score Kaiser : sinus 24 x 23 mm (Z score + 4.1), ST junction 18 x 18 mm (Z score + 3.2), ascending aorta 18 x 16 mm (Z score + 3.1), transvers arch 15 x 14 mm (Z score +4.6), isthmus 9 x 9 mm (Z score + 0.5), diaphragmatic aorta 9 x 8 mm (Z score + 0.6). The brachio-cephalic arteries were dilated and tortuous.
She repeated a CT angiogram three years later that showed findings in keeping with her previous one. Z score kaiser : Sinus 26 mm (Z score + 4.7), ST junction 18 x20 mm (Z score + 2.5), ascending aorta 19 x 19 mm (Z score + 2.4), transvers arch 11 x 13 mm (Z score -0.1), isthmus 12.5 x 12.5 mm (Z score + 0.9), diaphragmatic aorta 11 x 11 mm (Z score + 1.1). Figure 4, table 1.
MANAGEMENT
Despite expanding knowledge and increased understanding of rare and novel syndromic inherited aortic diseases management is largely extrapolated from more common connective tissue diseases with cardiovascular involvement and namely Marfan and Loeys-Dietz syndrome. According to current clinical practice she was started on beta-blockers to slow down the progression of the aortic growth. She started atenolol on a low dose that was then titrated up to 2 mg/Kg which she tolerated well. When she was 5 years old after repeating the CT angiogram Angiotensin II receptor blockers were added (losartan 0.6 mg/kg). A plan of cardiac regular surveillance is in place with six monthly echocardiograms and cross-sectional imaging every 12-18 months.
CONCLUSION
Cutis laxa syndrome is a rare connective tissue disorder that can present with a variety of clinical manifestations, including cardiovascular involvement. In this case report, we present the case of a child with ADCL with aortic dilatation documented in early infancy and with family history of severe aortic dilatation. The genetic proband in the family is our patient and no tissue was available for confirmation in the father who sadly passed away before her birth. While the phenotype is very variable in CL it is important to screen for cardiovascular involvement as the location and extent of vascular involvement can be significant and drive morbidity and mortality in a number of patients starting from a young age.
Early cardiovascular screening should be put in place for every patient with CL. Regular monitoring of aortic dimensions and other cardiac parameters can help identify and manage potential complications, such as aortic dissection or rupture. The prognosis for patients with CL can vary depending on the severity of the condition and genotype. While the patient in this case report has so far shown relatively stable aortic dimensions with medical therapy, the long-term prognosis remains uncertain, especially given the family history of severe aortic disease. Continued surveillance and early intervention are essential for optimizing the outcomes of patients with cutis laxa syndrome.