
Take-home messages
- Echo imaging that reveals LV hypertrophy may be indicative of cardiac amyloidosis (CA)
- GLS pattern of apical sparing is associated with CA
- Cardiac MRI can be very sensitive for the detection of myocardial infiltration but does not confirm the type of CA
- ECG can suggest cardiac involvement in CA but is rarely confirmatory
- Tissue biopsy, including myocardial biopsy, is usually necessary to establish the fibril type with CA
Introduction
Amyloidosis is a challenging multisystem condition that requires a high level of suspicion and careful evaluation of the patient history, physical examination, and available testing in order to make an early and accurate diagnosis, but also to define the extent, if any, of cardiac involvement. Depending on the manner of presentation, specific testing can effectively aid in detecting the presence of cardiac amyloidosis (CA). The multisystemic nature of this illness makes the condition difficult to treat in many situations, but certainly early identification and treatment are connected to the highest level of success [1-4]. Thankfully, many prospective clinical trials for the treatment of cardiac amyloidosis are underway and future therapies are very promising [5].
Since the presentation of this heterogeneous disease can be perplexing, cardiologists have to be aware of subtle findings and make clinical connections between seemingly disparate symptoms and signs in order to have the best overall outcome. A patient may present with any combination of cardiac, neurological, gastrointestinal, orthopaedic, or renal manifestations of amyloidosis, and typically these symptoms may be nonspecific. It has been recognised that providers may attribute symptoms to more common diseases without further investigation.
Once the identification and treatment of the amyloidosis is delayed, the benefit of targeted therapy for amyloidosis due to AL (light chain) or ATTR (transthyretin) is substantially reduced. It is important to note that a majority of educational efforts typically focus on a single organ system but the cornerstone of treatment of any form of amyloidosis, with or without CA, is a comprehensive multidisciplinary approach. The purpose of this review is to emphasise the practical opportunity for an early diagnosis of CA, keeping the multidisciplinary team approach in mind, and improving the amyloidosis-specific treatment as a result.
The landscape of treatment has changed dramatically in the last decade and guideline-type position statements are now available to provide a structure for diagnosis and treatment to clinicians [6,7]. While this is a tremendous development, the point of contact for a patient is commonly a routine visit to a provider with general symptoms that may be mild or not yet recognised as significant. The portal for entry into medical evaluation would likely involve standard electrocardiogram (ECG), echocardiography (echo) and laboratory testing. Since this is the introductory pathway for the diagnosis of CA, let’s describe “red flag” type initial findings that would warrant further evaluation with more advanced testing.
Red flag historical and physical findings
“Red flag” symptoms and physical findings may be present individually, or in combination, and if present should stimulate the provider to be inquisitive and consider advanced testing to discern if CA is present. Conditions that are highly suggestive of CA and would warrant further confirmation include bilateral carpal tunnel syndrome, with a biopsy done during the surgery to make the diagnosis [8]; unexplained sensorimotor peripheral neuropathy, autonomic dysfunction with accompanying orthostatic hypotension can be telltale findings in multiple subtypes of amyloidosis; and gastrointestinal symptoms such as unintentional weight loss, early satiety, nausea, diarrhea, and constipation can indicate multisystem involvement [9].
Imaging
Echocardiography (Echo)
The classic presentation of CA is one of heart failure with preserved left ventricular function (HFpEF). In fact, recent data suggest that this may occur more frequently than suspected, especially if one considers the underlying cause of heart failure and cardiomyopathy [10]. Since any initial evaluation for heart failure signs and symptoms would include cardiac biomarkers and Echo, this has become the typical entry point for the diagnosis of CA. However, routine Echo images in patients with CA may be nonspecific and overlap considerably with HFpEF (Figure 1 A, B, C). As a result, the potential delay can be up to 36 months before a definitive diagnosis of CA is made [11]. Transthoracic echocardiography is widely available and may be useful to differentiate cardiac amyloidosis from other causes of increased left ventricular wall thickness.
Figure 1. Selected classic images on Echo for cardiac amyloidosis. A. Parasternal long axis image of left ventricular hypertrophy. B. Short axis image of left ventricular hypertrophy. C. Bull’s eye display of left ventricular strain is consistent with apical sparing.
1A.
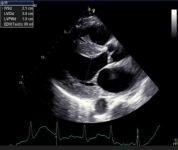
1B.
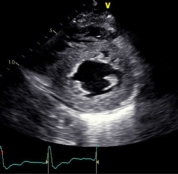
1C.
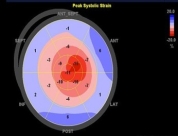
Global longitudinal strain (GLS) and apical sparing pattern have been useful in differentiating CA from other causes [12-14]. However, these parameters are not routinely measured in standard
echocardiographic laboratories, and their diagnostic performances have not been validated in populations with clinical suspicion for CA. In fact, recent data suggest that these tools are not specific to CA, but, if investigated completely when encountered, these parameters could lead to an earlier diagnosis of CA [15].
Cardiac magnetic resonance imaging
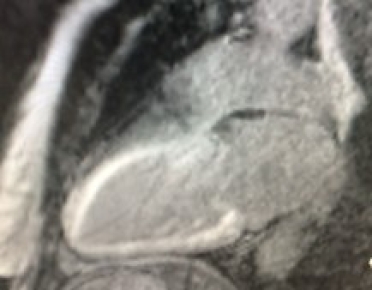
Cardiac magnetic resonance (CMR) imaging with gadolinium contrast can be useful in the identification of cardiac amyloidosis. Late gadolinium enhancement (LGE) occurs as contrast accumulates in the increased extracellular volume (ECV) secondary to amyloid deposition in the myocardium. LGE seen by CMR is one of the most accurate predictors of endomyocardial biopsy-positive amyloidosis [16]. There are a number of other CMR parameters that may be useful for the characterisation of myocardial tissue and possible infiltrative disease [17]. Recent data confirm the utility of the ECV value to assist in the diagnosis of CA [18]. CMR is a good early screening tool for cardiac amyloidosis and can be useful for detecting early fibrosis or infiltration prior to increasing LV thickness (Figure 2 A, B).
Figure 2. Common patterns of hyperenhancement on CMR potentially indicating cardiac amyloidosis. 2A. Diffuse hyperenhancement. 2B. Extensive subendocardial hyperenhancement on cardiac magnetic resonance imaging.
2A.
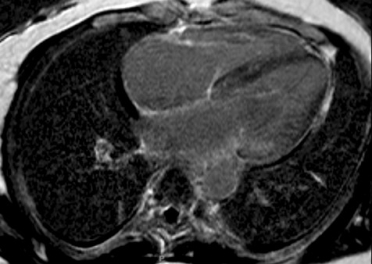
2B.
With equivocal cardiac imaging or if AL cardiac amyloidosis needs to be differentiated from TTR cardiac amyloidosis in the setting of plasma cell dyscrasia, an endomyocardial biopsy should be performed. Up to 25% of patients with WT-TTR cardiac amyloidosis will have a monoclonal protein [19]. In addition, a substantial percentage of cases of amyloid may have a different amyloid subtype (AL vs. TTR) than what was clinically suspected [20]. Therefore, definitive amyloid subtyping is critically important for therapeutic decision-making.
Electrocardiogram
The standard ECG can potentially indicate an infiltrative process but this test by itself will only rarely be enough to be confident of a diagnosis of CA [21]. The primary role an ECG plays in the evaluation of patients with potential CA is one of assessing for rhythm disturbances and conduction diseases that may require specific therapy. The classic description for CA is a mismatch between left ventricular hypertrophy (LVH) seen on imaging and low voltages seen on ECG (Figure 3 A, B). Unfortunately, this is not a common occurrence unless late-stage disease is present.
Figure 3. Common ECG manifestations of cardiac amyloidosis
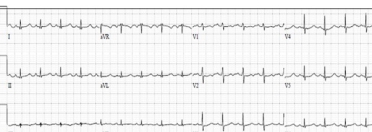
ECG #1. This 64-year-old patient had sinus tachycardia and diffusely low voltages throughout. Echo showed normal left ventricular ejection fraction (LVEF) with mild left ventricular hypertrophy (LVH) and a reduction in global longitudinal strain (-13.2%). This patient had light chain (AL) amyloidosis and was started on chemotherapy appropriate for that condition.
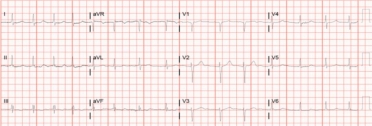
ECG #2. This 67-year-old patient had a 2-year history of atrial fibrillation with cardiac biomarker elevation (troponin I and NT-proBNP) on multiple occasions. LVEF was 50% with mild to moderate LVH and no evidence of coronary artery disease by angiography. Note the anterior infarction pattern and overall lower voltages in addition to the atrial fibrillation.
An ECG can be useful but not typically diagnostic of CA [22]. Since this is a routine basic test, it may serve as a tool to indicate the need for further evaluation in order to exclude CA.
Pyrophosphate scan
Radionuclide scans (Tc99m PYP; Tc99m DPD) are very useful for the diagnosis of CA related to ATTR (both the mutated and wild type). It is imperative to exclude AL CA prior to obtaining the radionuclide scan since the AL variant can lead to false positive results. Serum free light chain level testing and serum protein immunoelectrophoresis can effectively exclude AL prior to obtaining the scan. Isotopes including 99mTc-DPD (3,3-diphosphono-1,2-propanodiacarboxylic acid) and 99mTc-PYP have a high propensity to bind to TTR amyloid deposits over AL amyloid or other infiltrating disease. Therefore, it is useful in detecting TTR amyloidosis and may be more useful than CMR for this reason [16]. Finally, with regard to imaging, positron emission tomography (PET) and the use of tracers such as 18F florbetapir and 11-C Pittsburgh B compound (PiB) are potentially useful in identifying ATTR and AL cardiac amyloidosis [23].
Serum cardiac biomarkers, serum free light chains, and protein electrophoresis
The measurement of cardiac biomarkers is an essential tool for the diagnosis of CA [24]. Natriuretic peptides and troponins I and T have all been validated for defining cardiac involvement and assessing prognosis. There are other cardiac-related biomarkers that have shown promise in the diagnosis and prognosis of CA but these have not been validated across a wide spectrum of patients [25]. In all patients with suspected CA, these biomarkers should always be assessed [26]. Supportive serum testing that is also necessary includes serum free light chain testing to screen for a monoclonal protein and a protein electrophoresis to define the potential source of monoclonal gammopathy.
A practical guide on interpreting these serum protein elevations can assist the clinician as to the next best step in pursuing the diagnosis of CA [27]. At this point in any evaluation, a consultation with haematology is usually required and encouraged. A multidisciplinary team approach to CA is truly an imperative [7,28]. When the diagnosis of AL CA is made, this should be considered a cardiac emergency and aggressive treatment should be instituted as quickly as possible [4,29]. Careful coordination with haematology is therefore essential.
Genetic testing
In all patients with suspected or confirmed transthyretin amyloidosis (ATTR), genetic testing is required. There are several new therapies that have been tested in patients with cardiomyopathy and/or hereditary polyneuropathy that have a genetic basis for ATTR. In patients with phenotypic hypertrophic cardiomyopathy (HCM), it is also necessary to do genetic testing to exclude CA or potentially diagnose another cause of HCM [30]. In the case of targeted therapy for CA, there have been some very important studies recently that have meaningful impact on overall patient outcomes [31-33].
The field of developing therapies continues to expand for this condition [34]. As it turns out, genetic screening for CA generally only uncovers a mutation about 15% of the time [9]. There are many cases of amyloidosis that are not associated with genetic abnormalities but still have the same basic cardiac phenotype. In this manner, genetic testing is not the most complete or most accurate method to detect CA but is complementary to the constellation of a number of biomarker and imaging tests, as well as mass spectrometry to confirm the fibril type [35].
When to suspect cardiac amyloidosis?
A recent expert consensus was published that attempted to address the practical considerations and provided an overview of available testing and summarise treatment strategies. The introductory figure in this expert document summarizes the early diagnosis and treatment of CA [7].
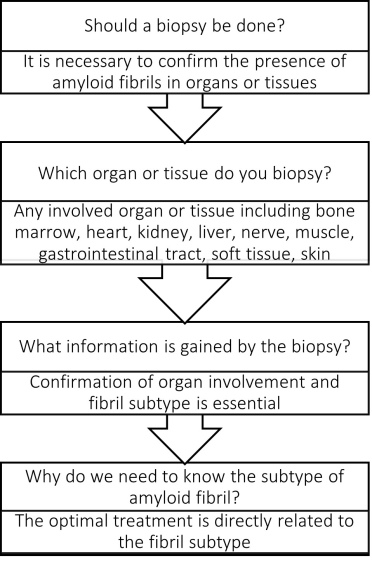
When is a biopsy necessary?
The gold standard for diagnosis of amyloidosis remains laser microdissection and tandem mass spectrometry (LMD/MS) from a Congo red-positive tissue biopsy [36]. A positive biopsy is indicative of amyloid in the heart, although very elderly patients may have patchy amyloid deposits that are not clinically significant. Cardiac biopsy may not be necessary if diagnosis can be confirmed from other tissue. Abdominal fat aspirate can be taken and Congo red staining has 70–90% sensitivity for diagnosis of systemic AL amyloidosis but is less useful in the diagnosis of ATTR amyloid [37].
Whenever there is equivocal cardiac imaging, or if AL CA needs to be differentiated from ATTR in the setting of plasma cell dyscrasia, an endomyocardial biopsy should be performed [7]. Therefore, definitive amyloid subtyping with LMD/MS is critically important for therapeutic decision-making regardless of the tissue from which it is obtained. A basic algorithm is proposed to help ascertain whether a cardiac biopsy is needed or if definitive information can be established from existing testing (Figure 4).
Figure 4. Basic algorithm to determine if a biopsy is needed for the diagnosis of cardiac amyloidosis.
Conclusion - Impact on daily practice
The accurate and timely diagnosis of cardiac amyloidosis (CA) requires a high index of suspicion and an appropriate response to certain clinical “red flags” which include a combination of historical and physical findings, cardiac biomarkers, as well as multimodal imaging. A definitive diagnosis can be made by a simple process in certain instances, but other presentations may require advanced testing that includes cardiac or other tissue biopsy with confirmatory testing typically only available at specialised centres. In light of the multisystem involvement and varying clinical presentation, a multidisciplinary approach is paramount. Biomarkers and imaging, along with specialised pathologic testing (mass spectrometry), are the cornerstones of diagnosis. There are many new therapies emerging that hopefully will have a major impact on the disease state and improve overall outcomes. The key element will always be early diagnosis and treatment. The future is very bright for patients that suffer with cardiac amyloidosis with these principles in mind.