


Take-Home Messages:
- Inherited Cardiac Conditions (ICCs) are an important cause of arrhythmias, especially in the younger population.
- Rare genetic variants are involved in the pathophysiology and clinical manifestations of ICCs leading to both brady- and tachy-arrhythmias.
- Genetic testing may help guide the diagnosis, treatment and risk stratification of several ICCs.
- Increasing evidence suggests a role of common genetic variants and polygenic risk in ICCs, although further research is needed before their routine utilisation in the arrhythmia clinic.
Introduction
Over 150 years ago the first theories on the mechanisms governing variability in phenotype between parents and offspring were made. Genetic technologies have evolved sufficiently to be able to uncover the whole human genome, the complete set of genes in a cell or organism [1]. Alongside this, our understanding of the mechanisms underlying the heritability of cardiovascular diseases has also progressed. Cardiac arrhythmias are no exception to this, and the role of genetic predisposition is increasingly being recognised.
Genetic technology
Next-generation sequencing
Current methods for clinical genetic testing (collectively known as next-generation sequencing [NGS]) employ rapid and large-scale analyses of panels of genes selected for association with the condition being tested for, or similar but ‘virtual’ panels from sequencing of the whole genome or the whole exome (the genetic code that directly impacts upon protein product). NGS data generated in research have resulted in large public databases and resources that characterise variation in the genetic code in the general population such as GnomAD (broadinstitute.org). These data, as well as information from the scientific literature, family and functional studies, have been incorporated into a framework for categorisation of rare genetic variants according to their likelihood of disease causation, i.e., their pathogenicity.
A consensus document from the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology published in 2015 [2] developed a five-tier system of classification for genetic variants relevant to Mendelian diseases based on several criteria, including: variant position, type and frequency in the general population; resultant changes in the transcribed mRNA sequence and predicted effect on translated protein function; inheritance mode and co-segregation in relatives; and association with clinical phenotype. Variants are ultimately classified as pathogenic (Class 5), likely pathogenic (Class 4), uncertain significance (VUS; Class 3), likely benign (Class 2), or benign (Class 1).
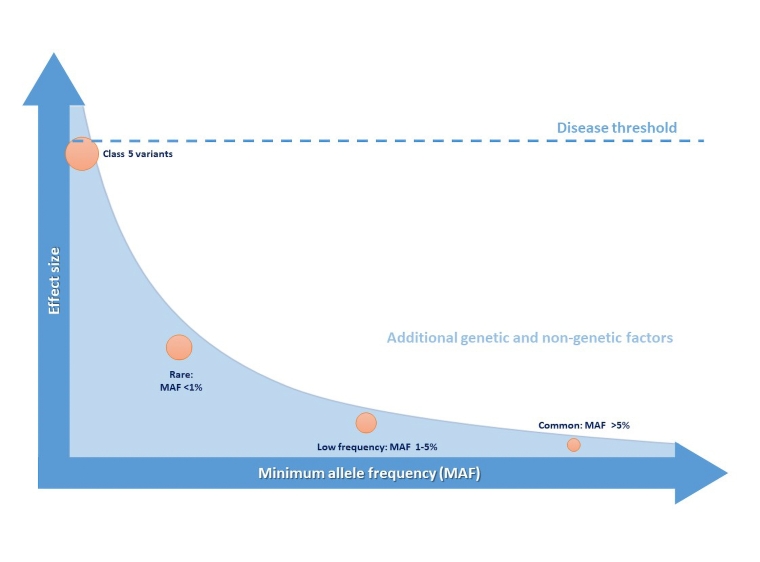
Most inheritable cardiac conditions (ICCs) largely behave as monogenic disorders, i.e., with a classical autosomal dominant Mendelian pattern of inheritance, such that there is 50% chance that the affected allele harbouring a Class 4 or 5 variant is transmitted to each child. These variants have a large effect size as they cause an important disruption in the biological processes associated with the development of the disease (Figure 1); however, the significance of a disease-causing variant in the clinical setting must be interpreted by also taking into account two phenomena: incomplete disease penetrance and variable expressivity. The former refers to the presence of individuals from the same family harbouring the same specific genetic variant as affected subjects, but that do not show evidence of the disease. The latter describes the difference in the clinical manifestation amongst subjects with the same genetic condition. Both disease penetrance and expression are believed to be influenced by non-genetic but also genetic factors as described below.
Figure 1. Genetic variant frequency and effect size and their role in pathogenicity for arrhythmic disorders including ICCs.
Polygenic heritability and disease susceptibility
Most of the causative genes for ICCs were identified using a candidate gene approach, either by genetic linkage analysis (based on the assumption that alleles localised closely on a chromosome tend to be inherited together) or biological pathway information. It has become evident that many ICCs are not entirely monogenic but, rather, have a complex genetic basis.
Genome-wide association studies (GWAS) have associated physiological traits or diseases with common genetic variants or single nucleotide polymorphisms (SNPs), defined as variants with a minimum minor allele frequency (MAF) in the general population of over 5%. GWAS compare the relative frequency of SNPs in affected and control populations, looking for genome-wide significant findings in an agnostic fashion, and uncovering new biological pathways as a consequence. However, the contribution of SNPs to disease development is usually much less significant than pathogenic rare variants while low-frequency variants (MAF 1-5%) may have intermediate effect sizes. In combination, however, SNPs and low frequency variants may contribute to or modify the risk of susceptibility to disease development, giving rise to a model of oligogenic or polygenic heritability (Figure 1). Indeed, the additive effects of permutations of multiple SNPs can be measured using polygenic risk scores (PRSs) that may have an important impact on phenotype and explain some of the “missing heritability” in otherwise Mendelian disorders, as well as variable expressivity [3].
Arrhythmias in the clinic and heritability of risk
Arrhythmogenic diseases may be secondary to micro- or macroscopic structural abnormalities (i.e., partial or total absence of structures, fatty and/or fibrous replacement of normal tissues, calcification) and/or be due to functional abnormalities of the action potential. Genetic predisposition to these pathophysiological changes may underlie tachy- or brady-arrhythmias observed in the clinic.
Indeed, the genetic susceptibility to cardiac arrhythmias has been documented following descriptions of familial clustering of the most common cardiac arrhythmias: atrial fibrillation (AF) [4], ventricular fibrillation (VF) [5] and progressive conduction disease [6]. AF is usually secondary to cardiovascular disease or other external factors; however, common and rare genetic variation has been associated with “lone” AF [4]. Sudden cardiac arrest (SCA) due to ST-elevation myocardial infarction survivors were found to have a significantly increased family history of sudden cardiac death (SCD) and SCA [5].
Ventricular tachycardia (VT) and VF are often a consequence of structural heart conditions, including cardiomyopathies that may have a strong genetic predisposition. Furthermore, the risk of SCD may be particularly high in young people and is also often associated with channelopathies [5]. Finally, bradycardia and heart block, commonly secondary to degenerative cardiac disease, may have genetic determinants and even appear from birth. Indeed, AF, VF and conduction defects may be the only manifestation of an ICC, isolated or in combination.
Whilst ICCs are typically caused by pathogenic rare variants, this monogenic model cannot explain all heritability underlying ICCs, and recent evidence supports the polygenic model, as described below [5].
Utility of genetic testing in the clinic
The prevalence of the most common rhythm abnormalities increases with age and comorbidities; SCA and SCD in the young most often result from inherited cardiomyopathies or channelopathies, although a high proportion remain unexplained [5]. International guidelines and expert consensus documents have been developed to guide the diagnosis and management of ICCs; however, the widespread integration of genetic testing into the clinical setting has forced clinicians and researchers to implement a rationalisation of not only variant interpretation but adjudication of genes suitable for such testing.
The National Institutes of Health Clinical Genome (ClinGen) Resource (clinicalgenome.org) has established multiple expert panels and a framework for undertaking this process in the ICCs. The ACMG framework for variant interpretation and the ClinGen recommendations have been included in the last 2022 EHRA/HRS/ APHRS/ LAHRS Expert Consensus Statement on the state of genetic testing for cardiac diseases [3]. Figure 2 summarises the diagnostic gene panels frequently associated with clinical arrhythmias and their associated phenotypes according to this latest consensus statement.
Figure 2. Classification of cardiac arrhythmias and overview of genes suitable for genetic testing according to ClinGen and the 2022 EHRA/HRS/ APHRS/ LAHRS Expert Consensus Statement on the state of genetic testing for cardiac disease. [3]
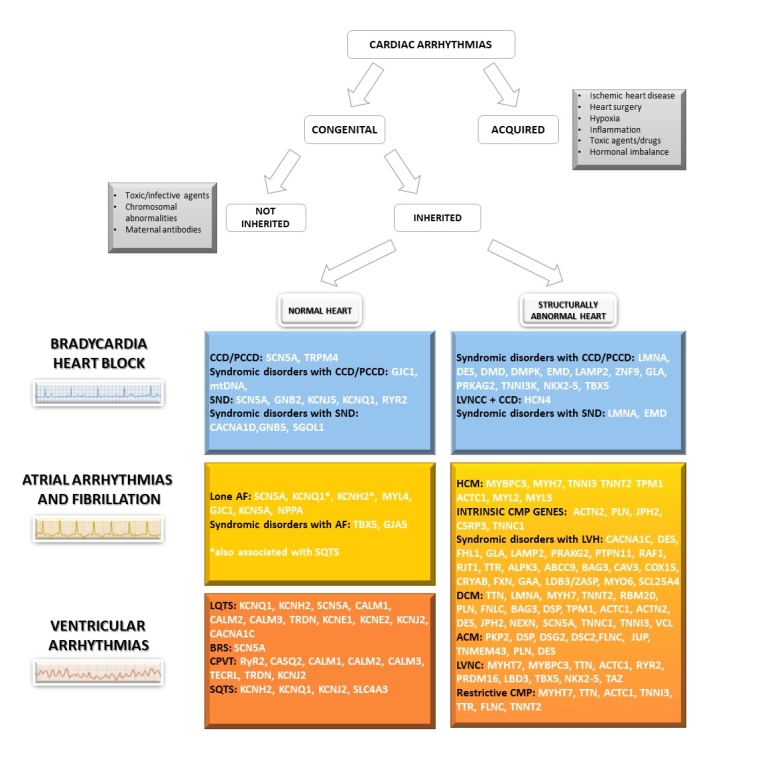
ACM: arrhythmogenic cardiomyopathy; AF: atrial fibrillation; BrS: Brugada syndrome; CCD: cardiac conduction disease; CMP: cardiomyopathy; CPVT: catecholaminergic polymorphic ventricular tachycardia; DCM: dilated cardiomyopathy; HCM: hypertrophic cardiomyopathy; LNCC: left ventricular non-compaction cardiomyopathy; LVH: left ventricular hypertrophy; PCCD: progressive cardiac conduction disease; LQTS: long QT syndrome; SQTS: short QT syndrome; SND: sinus node disease.
After the identification of index cases (probands), cardiologists and genetic counsellors must identify family members potentially at risk, through clinical screening of immediate relatives and/or predictive genetic testing, i.e., testing of Class 4 or 5 variants in relatives to make or exclude a diagnosis. In this context, a detailed three-generational family history may add supporting evidence of the phenotype in a family and improve interpretation of Class 3 variants through assessing segregation of phenotype with genotype. The genetic screening of families is particularly important in channelopathies such as long QT syndrome (LQTS), Brugada syndrome (BrS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and in the cardiomyopathies such as hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), and arrhythmogenic cardiomyopathy (ACM), where identification of a causative variant should be pursued even in the absence of symptoms or phenotype, to establish appropriate preventative measures.
Genomic risk
SCD/SCA
SCD in young individuals may be due to a spectrum of cardiac diseases with different arrhythmic risks, including channelopathies, cardiomyopathies, congenital heart disease, myocarditis, and premature coronary artery disease. Therefore, post-mortem evaluation, ideally including a toxicological examination and an expert pathology review, is of utmost importance to investigate any case of sudden unexpected death.
Results from a recent European Heart Rhythm Association (Heart Rhythma Associaton EHRA) survey have shown, however, that an autopsy is performed in only 43% of sudden unexplained deaths, a macroscopic examination of the body and all organs in 71%, and expert cardiac examination in 32% of autopsy cases [7]. Similarly, post-mortem genetic testing (molecular autopsy), which proved to increase the yield of diagnosis in ICC families [8] and is advocated by current guidelines, is requested in only 37% of cases [7]. A study using whole exome sequencing has identified ACMG Class 4 or 5 variants in genes associated with coronary artery disease, cardiomyopathies, channelopathies and aortic disorders in up to 2.5% of unselected cases of SCD [9]. The contribution of common genetic variation to SCD is supported by one study associating the 2q24.2 locus, containing 3 genes with unknown function significance, with an increased risk of SCD [10].
Recent data from the CASPER registry, a prospective Canadian national registry of cases of initially unexplained SCA, showed that 10% of survivors carry a disease-causing variant in genes associated with ICCs, mainly associated with cardiomyopathy [11]. This is in keeping with a prior study highlighting a genetic yield of ACMG Class 4 or 5 variants in established SCA-predisposing genes ranging between 2% and 17%, depending on the type of genetic test utilised (e.g., disease-specific vs whole exome sequencing) in subjects without discernible cardiac phenotype (idiopathic VF) [12]. Moreover, the possibility of detecting ≥1 Class 3 variant in channelopathy or cardiomyopathy genes (15-26%) exceeded that of clinically actionable Class 4 and 5 variants (2-17%), challenging the utility of pan-arrhythmia, pan-cardiac, and exome-based genetic testing in unexplained SCA survivors with a diagnosis of IVF.
A founder variant, a pathogenic variant with a relatively high prevalence in the population in a particular geographical region, derived from a common ancestor, and whose presence is immediately useful as a diagnostic risk marker, has been associated with an increased risk of sudden death: the DPP6 haplotype on chromosome 7q36 in idiopathic VF survivors of Dutch ancestry [13]. So far common genetic variation has not been associated with SCA. Thus, current guidelines do not support hypotheses-free genetic testing using whole exome or genome sequencing in survivors of unexplained SCA, although testing channelopathy and cardiomyopathy genes may be useful, and in selected cases, testing for founder variants may also be of utility [3].
AF
Rare monogenic forms of familial AF (Figure 2) have been described as have associations with common genetic variation. Indeed, more than one hundred SNPs have been associated with ‘lone’ AF, identified mainly in non-coding regions of the genome [3, 14]. In the ICC clinic, AF can be associated with both primary arrhythmia syndromes and cardiomyopathies, but familial monogenic AF has been observed in up to 5% of all AF cases referred to an arrhythmia clinic [4]. Current evidence supports a potential role of genetic testing in index patients with an established diagnosis of familial (age <60) AF [3].
Genomic risk in ICC-related arrhythmias
As mentioned above, rare genetic variants in single genes with large-effect size usually underlie the development and clinical manifestations of ICCs (Figure 1). In addition, a significant shift from the concept of “one gene - one disease” has occurred, with several examples of a single gene affecting multiple phenotypes (Figure 2). Due to the high clinical and genetic variability of ICCs, careful risk stratification approaches must be adopted, and therapeutic strategies tailored to the individual. Depending on the disease, genetic testing results may have a significant impact not only on the diagnosis, but also in the risk assessment and therapeutic approach [3].
In patients with LQTS the risk of life-threatening arrhythmic events is based on the genetic substrate and is modulated by the QTc measurement; specific genes and variants are associated with different arrhythmic risk and potential therapeutic benefits. For example, the LQT2 and LQT3 subtypes, encoded by KCNH2 and SCN5A genes respectively, carry higher risk for life threatening arrhythmic events than the LQT1 subtype, encoded by KCNQ1. This has been characterised in a few different risk models including a recent risk calculator, 1-2-3-LQTS-Risk, developed in an Italian population and validated in the international LQTS Registry [15]. Furthermore, medical treatment with betablockers is recommended in patients with a pathogenic LQTS variant, regardless of the degree of QTc prolongation, while mexiletine should be considered in patients with LQT3. Similarly, patients with RYR2 variants but limited or absent manifestations of CPVT should also be considered for medical therapy. The identification of SCN5A gene variants in subjects with BrS may be associated with worse prognosis or overlapping cardiac conduction disease, therefore requiring preventative device implantation.
Patients with desmosomal variants causing ACM should avoid competitive sport or high-intensity exercise, which can worsen ventricular dysfunction and increase the incidence of ventricular arrhythmias. Dilated cardiomyopathy patients who carry pathogenic variants in genes associated with higher risk of SCD such as LMNA, PLN, DSP, FLNC or RBM20, may be considered for preventative PM/ICD implantation, a higher ejection fraction (<45%) than usually employed in non-genetic cases (<35%) [16].
LMNA cases are also substantially at higher risk of developing bradyarrhythmias and may warrant pacing at an earlier stage and thus may be considered for device therapy at an earlier stage. In some syndromic conditions associated with left ventricular hypertrophy which mimics HCM, the identification of a specific genotype may prompt potential disease modifying treatment such as enzyme replacement therapy in patients with GLA variants and Fabry’s disease. Finally, one study demonstrated the feasibility of suppression-and-replacement hybrid gene therapy in an in vitro model of LQT1, thus laying the foundations for a future therapeutic approach to ICCs, directly targeting the abnormal substrate [17].
Aside from the effect of monogenic rare variations, arrhythmic manifestations of ICCs may be influenced by the presence of one or multiple common SNPs in the genome, contributing directly to disease causation. This has been firstly investigated in the BrS, where only 20% of cases exhibit a Class 4 or 5 SCN5A variant and heritability remained largely unexplained. A first GWAS, performed on approximately 300 BrS probands and 1000 control subjects, led to the identification of three significant loci associated with the disease at the SCN5A, SCN10A, and HEY2 genes [18]. Other loci have been identified in subsequent studies. A significant burden of common variation associated with the QT interval is recognised, with a PRS derived from GWAS of the QT interval in the general population able to explain in part the heritability of LQTS in patients compared to healthy people; most significantly in those without an ACMG Class 4 or 5 genetic variant [19].
Alternatively, SNPs may act as modifiers of phenotype or acquired disease. For instance, a PRS derived from the first BrS GWAS can predict the occurrence of the type 1 Brugada pattern in response to the ajmaline provocation test. Common variants can also explain in part incomplete penetrance of the Brugada phenotype in families with Class 4 or 5 SCN5A variants [20]. In addition, the QT interval PRS may predict the individual susceptibility to drug-induced QT prolongation and Torsades de Pointes [5]. Although promising as diagnostic or prognostic tools, these are not currently adopted in the clinical setting [3].
Conclusion
Recent technological advances and developments in our understanding in the genetic architecture of heart disease are leading the way towards new diagnostic and therapeutic approaches to arrhythmias. Genomic techniques generate enormous amounts of data, which are currently not yet used to their fullest potential, due to analytical and interpretative challenges. Furthermore, the challenge remains to complete the shift from disease association to causation to make relevant conclusions about genomic-derived risk of arrhythmias. Currently, the applications of genomics include the early identification of index subjects and their relatives at risk of an ICC, the guidance of more individualised therapeutic approaches and prevention of SCD.