Introduction
A more recently described cause of LQTS, termed "calmodulinopathies", encompasses a spectrum of genetic arrhythmic syndromes arising from variants in CALM1, CALM2, and CALM3 genes4–6. These genes, located on distinct chromosomes (chr14q31, chr2p21, chr19q13, respectively), exhibit differential expression patterns across cardiac cells. Despite this divergence, they encode for the same protein known as CaM, a 149-amino acid calcium-modulated protein crucial for downstream signalling processes7.
Amongst others, CaM is tethered to the pre-IQ motif of L-type calcium channels (ICaL) (Image 1) and to the slow component of the delayed-rectifier potassium channels (IKs), modulating their function by either stimulating or inhibiting them. Variants in CaM cause a gain of function of these inward current channels, resulting in prolongation of the QTc8,9.
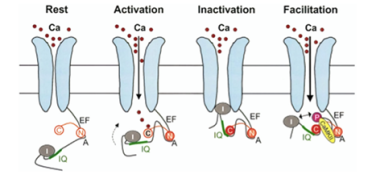
In terms of phenotype, this has been reported to be diverse, from channelopathy to syndromic forms and from early onset of life-threatening arrhythmias to the absence of symptoms. Whilst rarer than variants in the K+ and Na+ channel genes, calmodulinopathies are important for clinicians to recognise early on due to the dramatic presentation of malignant, and often lethal, arrhythmias in infants and children10,11.
Here we discuss the case of an infant exhibiting early-onset arrhythmogenic episodes, leading to a diagnosis of LQTS which was, through repeated genetic testing, eventually linked to a pathogenic CALM1 gene variant. This case is used to demonstrate the acute presentation of calmodulinopathies, the importance of a multidisciplinary, multicentre and specialist approach, as well as the possible phenotypic evolution of patients with calmodulinopathies, making the molecular diagnosis important for personalised treatment.
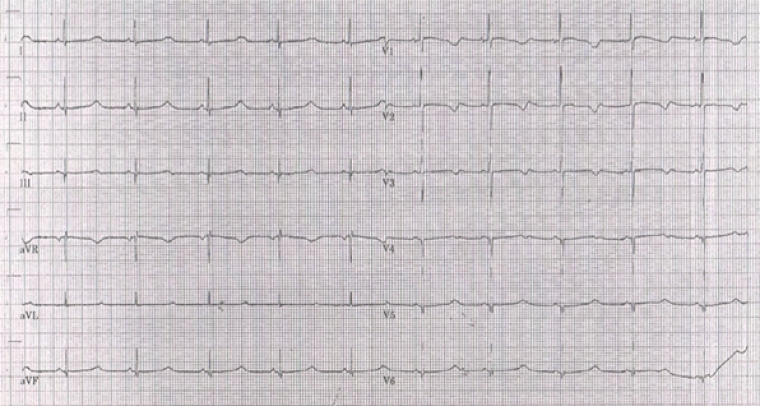
A previously well one-month-old infant boy from Malta manifested jerking movements and cyanosis of the lips during feeding, subsequently experiencing a brief episode of unresponsiveness lasting approximately 2-3 minutes. The parents administered a few rescue breaths, resulting in the infant's recovery. Following this episode, the patient was admitted to their local hospital, where an electrocardiogram (ECG) was obtained (image 2). Due to the corrected QT interval (QTc) being calculated at 500 milliseconds (ms) in lead II, a diagnosis of LQTS was made. The patient was commenced on propranolol, discharged home and referred to a cardiologist. The patient remained under close follow up and propranolol was up-titrated, as allowed by the resting heart rate, reaching a maximum dose of 1.8mg/kg/day. Past medical history was negative, except for reported history of slow fetal heart rate which was not investigated.

During follow up, a second episode occurred at 1.5 years of age when the infant was upset. As he was crying, his father picked him up and he started developing jerking movements of the body and developed cyanotic lips. He became limp and unresponsive, at which point his parents gave some rescue breaths and he recovered. This episode lasted a total of roughly two minutes. The local hospital referred the case to our unit for consultation regarding ongoing management, given the patient’s ongoing episodes, despite treatment on propranolol and having reached a QTc of 600ms (image 2). Given the presence of abnormal QT prolongation and suspected arrhythmogenic in nature episodes, a consensus was reached for the implantation of an implantable cardioverter-defibrillator (ICD) and subsequently an epicardial, single-chamber device was successfully fitted in our unit. The patient was discharged home on mexiletine (5mg/kg/day) and propranolol (1.75mg/kg/day).
As the patient developed peripheral oedema after introducing mexiletine, leading to his hospitalisation, this medication was discontinued by the local medical team. Over the following six months, two episodes of loss of consciousness were documented, with ventricular tachycardia/ventricular fibrillation detected and appropriately treated with ICD shocks.
The initial genetic panel sent at 2 years of age, which consisted of 5 causative genes for LQT 1 to 5 (KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2), failed to yield a result. After seeking a second opinion, from a specialist centre in Italy, a trio exome analysis was conducted, revealing a de novo pathogenic variant in CALM1—specifically, E141V. This variant alters the second nucleotide of CALM1 codon 141. Importantly, this variant was not found in the Genome Aggregation Database (gnomAD). Functional studies were then conducted and revealed reduced Ca2+ binding affinity. Additionally, channel inactivation was slower and less complete, with significant differences in peak and late Ca2+ currents compared to the wild-type (WT) form.
Two years post-implant, when the patient was 3.5 years old, the patient's device exhibited oversensing of T waves exclusively on paced QRS complexes, inhibiting pacing and resulting in an overnight heart rate drop to 40 bpm. Increasing the sensing threshold to 0.9mV did not resolve the issue. Consequently, the patient was considered for defibrillation threshold (DFT) testing and device revision.
During the procedure, VF was induced with burst pacing after administering Isoprenaline. Despite adjusting the sensing threshold to 0.6mV, the ICD could not effectively detect VF, and a successful shock required changing the configuration from coil to can. Recognizing the device's inability to safely provide effective therapy during VF, a multi-disciplinary team meeting approved a "hybrid approach". The patient underwent ICD system revision, with the generator changed and left in an abdominal position. A transvenous atrial and ventricular pace-sense lead was inserted and tunnelled subcutaneously to the abdominal pocket, and an ICD shock coil was positioned subcutaneously anteriorly at the left parasternal site, with the shock set between the prior epicardial coil and the new subcutaneous coil (Image 3, 4a,b).
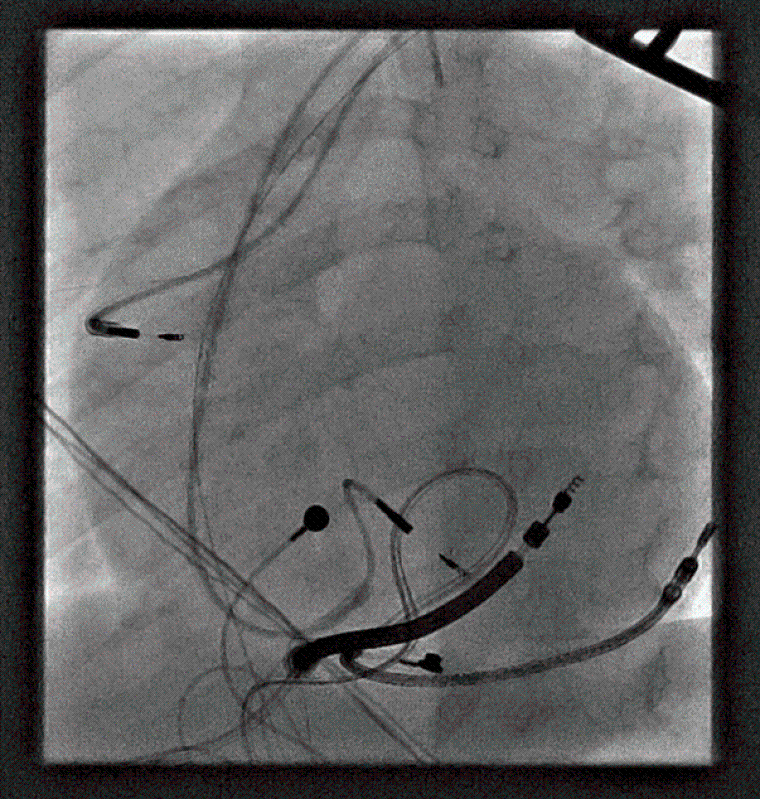


Left cardiac sympathetic denervation was contemplated, as this is often utilised in LQTS patients at high risk of recurrent ventricular fibrillation and sudden cardiac death, more so when medical treatment options fail to prevent these from happening12,13. Despite the documented benefits and following discussion at a multidisciplinary meeting for this child, this procedure was postponed due to the absence of subsequent events.
At the age of 7, the child experienced a third appropriate shock for ventricular fibrillation (VF), having been episode free since 2014.
At the age of 4 years, the patient started developing a distinct echocardiographic phenotype (Image 5), with a dilated left atrium (LA) (LA area 12.1 cm3, z score +5.7) and right atrium (RA) (RA area 10.8 cm3, z score +4.1), preserved global left ventricular systolic function, but impaired longitudinal function (tissue doppler lateral S’ wave 0.07m/s, septal 0.05m/s) and evidence of early impaired left ventricular relaxation (isovolumetric relaxation time 114ms, pulmonary vein A-wave duration 166msm abnormal mitral valve doppler). The left ventricle itself was normal in size, with a maximal wall thickness of 5-6mm (z score +02), with no significant valvular or other abnormalities.

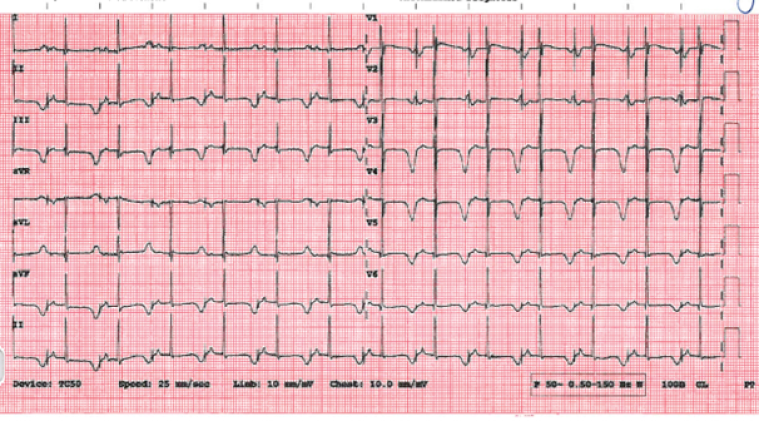
The patient is now 11 years old. He is seen locally as well as by our service, and remains on 2mg/kg/day of nadolol with no further documented arrhythmic events. He is mainly paced in the atrium (99.7%) from his device. His ECG (Image 6) and echocardiogram (Image 7) remains stable.
As the patient grew older, he became increasingly aware of the presence of his ICD. He was therefore referred and reviewed by our unit’s ICD Nurse Practitioner, to provide him with added support leading to better understanding the implications of living indefinitely with this device. Psychology support was also offered to the patient and the family.
Also, with increasing age, extra-cardiac manifestations of his diagnosis became apparent. He has been diagnosed with neurodevelopmental delay, learning difficulties, difficulties with concentration, and dyslexia.
Discussion:
This case stands out for several reasons.
Firstly, it draws attention on the importance of having an open mind when approaching patients presenting with unresponsive or syncopal episodes. Inherited channelopathies have been a longstanding cause of such type of presentation, but, especially in the neonatal period, brief unexplained episodes can be attributed to an intercurrent illness, respiratory or neurological cause as well. Taking appropriate history of the event and thinking wider about its aetiology, which includes performing an ECG as part of the assessment, is pivotal when making differential diagnosis.
It emphasises the importance of considering causative pathogenic mechanisms beyond the well-known K+ and Na+ channel variants in Long QT Syndrome (LQTS). The effects of variants in the calmodulin genes, due to the protein’s complex interactions, extend beyond purely arrhythmic manifestations, indicating a broader range of clinical features that should be considered in diagnosis and management10,11.
Medical professionals should not limit their focus to established pathways but remain open to exploring alternative causes and mechanisms. This aids in providing appropriate management to these patients, which is crucial in LQTS, as the severity of their phenotype is different according to their specific genetic variant14,15. Families benefit greatly from identifying pathogenic variants in probands, as this allows for cascade screening and risk stratification of first-degree family members, preventing them from presenting later in life with ventricular arrhythmias and sudden cardiac death 16.
It is vital that the psychological wellbeing of children and their families is constantly re-evaluated, and that multidisciplinary approach is always used when their presentation is early in life and dramatic, such as with an out-of-hospital cardiac arrest. Cardiovascular and psychological expertise should go hand in hand in ensuring that these patients and their families’ needs are fully met in all aspects of their life. Expert counselling from multiple professionals (such as specialist cardiologists, genetic counsellors, nurse specialists and clinical psychologists in paediatric services) can effectively assess and tackle distress and anxiety in children and their families.17–19
There are no doubts that multicentre studies have always been crucial in scientific research for data gathering purposes. Whenever new and rare conditions with limited literature and guidelines such as calmodulinopathies are first documented, registries like the ICalmR, whose entries are structured and handled by experts from multiple centres, provide an excellent source for critical knowledge in treatment and management of the aforementioned diseases10.
Ultimately, this case challenges the outdated notion that channelopathies, including LQTS, only manifest later in life. Early presentation in paediatric cases underscores the need for increased attention to paediatric services in understanding, diagnosing, and managing such conditions.
Conclusion:
Since its first description, the aetiology of LQTS is not limited to potassium-channel diseases, as more and more research works describe the pathogenic role of calmodulin variants.
This case highlights the complexity of LQTS, encourages a broader exploration of causative mechanisms, emphasises the syndromic nature of certain mutations, challenges preconceived notions about disease presentation, and underscores the importance of a collaborative, multicentre approach in the ever-changing field of genomic medicine.