Background
Mitral valve prolapse (MVP) complicated with arrhythmia is an emerging distinct entity, known as “arrhythmic MVP”(1). The clinical phenotype as well as the burden of arrhythmia are highly variable, with both atrial and ventricular arrhythmia being described as part of the arrhythmic MVP. Furthermore, a certain subset of patients with MVP were found to be at a higher risk of sudden cardiac death (SCD) due to ventricular arrhythmia (VA), independent of the degree of mitral regurgitation (MR) and left ventricular systolic function (2, 3). However, the relationship between MVP, VA and SCD is not entirely understood. Several features, such as impaired ventricular mechanics due to abnormal tethering forces, myocardial fibrosis, and association of mitral annular disjunction (MAD), might be helpful in identifying patients at risk (1, 3) Certain genes are associated with MVP, such as FILAMIN A (FLNA) and DACHSOSUS1 (DCHS1)(1). In clinical practice, routine genetic testing is not justified and is limited to patients with syndromic diseases (Marfan and Loeys Dietz syndrome)(1). The description of a number of pathogenic (P) or likely-pathogenic (LP) variants associated with cardiomyopathies and/or channelopathies in patients with MVP raises the question if this overlap contributes to the development of arrhythmic events and SCD (4, 5).
Patient presentation
We present the case of a 40-year-old female patient who experienced sudden cardiac arrest at 36 y.o. due to ventricular fibrillation (VF). Her ECG showed flattened T waves in the lateral leads, while bi-leaflet MVP and preserved left ventricular ejection fraction (LVEF) were documented at the transthoracic echocardiography (TTE). She received an ICD in secondary prevention. The ICD was interrogated 6 months after, revealing numerous monomorphic non-sustained ventricular tachycardia (NSVT), despite treatment with amiodarone. She was referred to an electrophysiology (EP) study. VT was induced after stimulation of the basal lateral left ventricular wall, 1 cm under the mitral annulus. Radiofrequency ablation (RFA) was successfully performed. The patient was referred to our center for second opinion, reporting no cardiac symptoms.
Diagnostic work-up
ECG showed sinus rhythm, with a slurred S wave in inferior leads and slurred R wave in lateral leads, as well as flattened T waves in aVL, with corrected QT of 460ms. No significant ventricular arrhythmic events were noted at the current ICD interrogation.

TTE was performed. The LV had a normal size, however, left ventricular ejection fraction (LVEF) was mildly impaired (48%) with mild longitudinal dysfunction (GLS -16,6%). The mitral valve appeared thickened and redundant, with bi-leaflet prolapse and mild regurgitation. MAD was seen, with a length of 4-5mm measured in mid-systole.
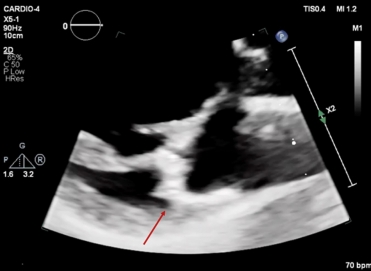

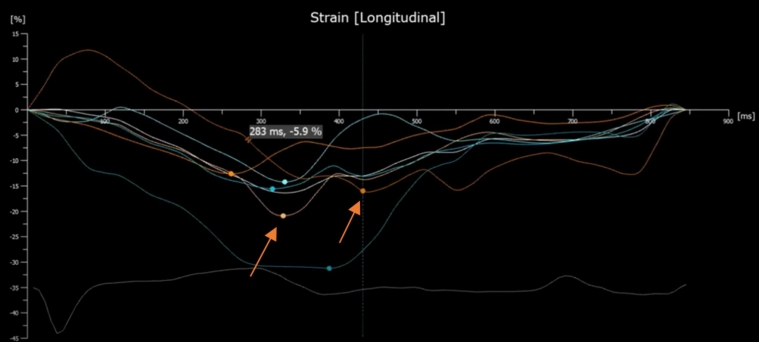
Tissue Doppler imaging (TDI) of the lateral wall revealed a second systolic peak, with a velocity of 17cm/s. Through speckle-tracking analysis, a double peak strain pattern is seen on the basal infero-lateral wall (orange arrows).

The patient’s family history was highly suspicious for an inherited cardiac disease, as her father died suddenly at 66 y.o. Furthermore, a paternal cousin required permanent pacing for a high-degree AV block at 25 y.o. and died suddenly at 30 y.o, while another paternal cousin had an aborted cardiac arrest at 21 y.o. Genetic testing was performed after genetic counselling, revealing a truncating (tv) likely-pathogenic (LP) Titin variant (TTN c.85150C>T. p.(Arg28384*) in a heterozygous form.

Cascade genetic testing was then proposed to the sister and surviving cousin.
Discussions
This is the case of a young female who experienced sudden cardiac arrest due to VF, initially believed to be secondary to abnormalities of the mitral apparatus (bi-leaflet MVP and MAD), as the subsequent EP study demonstrated inducible VT in the proximity of the mitral annulus. However, as the follow-up TTE showed mildly impaired LVEF and GLS, and considering the patient’s family history of sudden and aborted cardiac deaths, an heritable cardiomyopathy was also suspected and next-generation sequencing (NGS) for dilated cardiomyopathy was performed. As a result, the patient was also diagnosed with hypokinetic non-dilated cardiomyopathy (HNDC).
It is difficult to try and estimate the individual contribution of the two entities (MVP and TTNtv) in the development of malignant VA and SCD in this case. The available literature discussing the overlap between MVP and cardiomyopathies is currently limited. So far, mutations in DMD, TTN, LMNA, FLNC and HCN4 have been described in different cohorts of patients with MVP (5-7). The association between cardiomyopathy related genes, MVP and SCD is yet to be confirmed, even if limited data show that LP/P mutations are more likely to be detected in young individuals with sudden unexplained death and MVP, than in those without MVP(5).
In this case, the TTN mutation is a truncating variant, meaning it leads to structurally impaired Titin. TTNtv affecting the A-band are autosomal dominant and are an established cause of dilated cardiomyopathy (DCM). Patients with DCM caused by a TTNv have a higher rate of ventricular tachycardia (VT), as opposed to patients without, with an estimated prevalence of VT of 64%, compared to only 21% (8).
On the other hand, regarding the MVP, the patient exhibits several risk features, such as:
- MAD, which is measurable in PLAX, in mid-systole. CMR can completely assess the extent of MAD, however this was not possible in this case, as the patient’s ICD was not CMR compatible (1). CMR would have also been helpful in detecting late gadolinium enhancement (LGE), which is a known risk factor VA in this setting.
- Bi-leaflet prolapse with redundant leaflets. However, the valves were not severely degenerated or thickened (1).
- Double-peak strain pattern of the basal infero-lateral wall. Recently, double-peak strain showed incremental risk of arrhythmia over fibrosis in patients with MVP, highlighting the interplay between abnormal LV mechanics and arrhythmic events (9).
- Inducibility of sustained VA may indicate a more diffuse myopathic process. Long-term success rates for VA ablation in MVP range between 60 and 84% and the outcome of procedure is not adversely impacted by the presence of MVP(1).
Conclusion
The interplay between cardiomyopathies and MVP, as well as the impact of the overlap between the 2 entities regarding outcomes and prognosis, are yet to be defined and established. The current clinical case features the overlap between a LP TTNtv and MVP as an extreme clinical presentation: aborted sudden cardiac death. Even if the abnormalities of the mitral apparatus were the first to be noted, with the development of left ventricular systolic impairment in the context of a highly suspicious family history, a cardiomyopathy was also sought. However, a causal relationship between TTN truncating variants and MVP has not been proven. Although genetic testing is not routinely recommended in patients with MVP, it should be performed in selected cases, when red flags suggestive of an associated cardiomyopathy or channelopathy are present, as this will impact the prognosis of patients and their families.