

Mitral valve prolapse (MVP) is a frequent disorder affecting more than 2-3 % of the adult population. Being a common disease, it appears for the vast majority of patients as sporadic, with up to 26% of cases presenting with a positive familial history, some of them being associated with syndromes such as aneuploidy, Loeys-Dietz, and aneurysm-osteoarthritis syndromes (among other syndromes related to TGF-b signalling pathway).1,2 Besides those syndromic entities, variants in FLNA, DCHS1 and DZIP1 provide a molecular explanation in only a few familial cases, highlighting the complex genetic architecture of MVP and the broad landscape of unexplained heritability so far.2,3 In the case of sporadic MVP, the molecular determinants underlying MVP progression remain poorly understood, and the contribution of a genetic component in modulating disease threshold and severity still needs to be defined. Why do some patients do develop MVP, and others not? Why do some of them present with evolutive mitral valve disease and evolve towards severe mitral regurgitation? Why do some develop heart failure, atrial fibrillation and sudden cardiac death? What is behind this huge clinical diversity?
Aiming at identifying the molecular determinants of MVP and the genetic risk factors for disease progression and severity, Roselli and colleagues performed the largest genetic association study for MVP by combining data from six international cohorts/biobanks, including the FHS, the Mass General Brigham (MGB) Biobank, the UK Biobank, the Broad Cardiovascular Disease Initiative (Broad CVDi), the MVP-FRANCE/ Three-City Study (3C), and the MVP-NANTES/D.E.S.I.R..4 By this approach the authors reached to genetically compare 4884 MVP cases with 434649 controls. Performing a primary GWAS study analysis, they identified 16 loci that are associated with MVP, harbouring common alleles (with minor allele frequency of 4-50%). Among those loci, seven of them were independently associated with patient outcomes, including the requirement for valvular surgery. In another sensitivity analysis, a genetic correlation was also identified with atrial fibrillation and non-ischemic cardiomyopathy being associated with defined genetic backgrounds.
Interestingly, the authors crossed the GWAS-identified loci with RNA-sequencing data from the mitral valve and cardiac tissues of normal mice. By this approach, they identified 20 candidate genes located at the identified loci such as Sec11A, Cand2, Sptbn1, Mzt1, and Lmcd1 (mostly expressed at the MV tissue), and Cand2, Alpk3, Rbm20, Tgfb2, and Atxn2 (expressed in cardiac tissue), highlighting an overlap between MVP and genes involved in TGF-b signalling pathway, and with cardiomyopathy, among others. A similar approach was performed at the protein level with human mitral valve tissue obtained from five healthy donors. By this approach, they identified five “MVP GWAS genes” with the highest detected protein levels in the human mitral valve: LTBP2, SPTBN1, LMCD1, BAG3, and SEC11A. Those results identified a set of candidate genes that could be involved in MVP. To develop new tools that could improve risk stratification, the authors developed a polygenic risk score (PRS) for MVP based on their broad GWAS results. Increasing the robustness of PRS by considering the added value of clinical factors (age, sex, hypertension, heart failure, diabetes, and myocardial infarction), they achieved a prediction model that reached a stratification power with an AUC of 0,677.
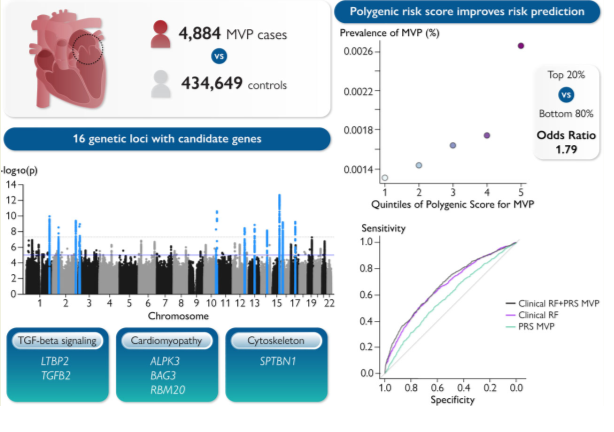
Graphical abstract: This study meta-analyzed 4884 mitral valve prolapse (MVP) cases versus 434 649 controls, and discovered 16 genetic loci associated with MVP. Downstream analyses implicated candidate genes involved in TGF-beta signalling, cardiomyopathy and the cytoskeleton. The results from the meta-analysis were used to calculate a polygenic risk score (PRS) to aid prediction of MVP. Adding the PRS to a model with age, sex and clinical risk factors improved MVP risk prediction. Abbreviations, MVP, mitral valve prolapse, p, p-value, PRS, polygenic risk score, RF, risk factors. Reproduced from Roselli et al.4, with permission.
Overall, this study leverages the contribution of genetic determinants in MVP, a disease which appears as sporadic for many cases, but with an overlooked familial dimension. Also, this study provides clues about the genetic background that could modulate disease severity and outcome. Through this very elegant study, the authors broaden the spectrum of genetic determinants underlying mitral valve disease and offer deeper insights into the pathological process leading to MVP. In particular, this study reinforces the overlap with other conditions such as aortopathies or cardiomyopathies: as examples LTBP2 and TGFB2 are both directly involved in TGF- signalling (involved in MVP, and familial thoracic aneurysms); the association with ALPK3 raise the existence of an overlooked overlap between cardiomyopathies and mitral valve disease; the association with BAG3 and RBM20 highlights an overlap with dilated and arrhythmogenic cardiomyopathy. In that view, MVP should no longer been seen as an isolated disorder, but should rather be considered as a component of a generally more systemic disorder overlapping with cardiomyopathies, arrhythmogenic disorders, connective tissue, or systemic diseases.5,6 This study supports a complex genetic architecture behind MVP and identifies new molecular candidates for the development of targeted therapies that could modulate MVP disease’s course and severity. Also, the long period between the detection of mitral valve disease and surgery should be seen as a missed opportunity to identify those disease determinants. Finally, this study highlights that genetics is no longer a matter of (rare) mono- or oligogenic disorders, but rather expands towards common alleles: complex modes of inheritance and PRS progressively broaden the spectrum of clinical genetics, providing a molecular basis for common diseases (with an interesting overlap in the identified molecular determinants with rare conditions). How those common variants will influence patient’s management at the single-person level still needs to be defined, but accumulating evidence reinforces that PRS will potentially change our way of assessing risk stratification and patient management, even in common diseases.